|
|
SVMalign Version V2.00 Authors: Thorsten Joachims <thorsten (a) joachims.org> & Philip Zigoris <paz3 (a) cs.cornell.edu> |
|

This document is organized as follows:
- Introduction
- Downloading and installing
- Model file format
- Input file format
- Program usage
- References
Introduction
In sequence alignment, one often has a good idea, a priori, of the appropriate values for various operations. For instance, your knowledge of bio-chemistry might tell you that the probability of a match occurring is greater than that of a protein being deleted from a sequence. Given these costs, one can find, by dynamic programming, the optimal sequence of operations that aligns two sequences. The problem of Inverse Sequence Alignment is, appropriately enough, the inverse of this. The learning problem can be generally phrased as, given a collection of sequences with pairs labeled either ‘similar’ (native and homolog sequence) or ‘dissimilar’ (native and decoy sequence), find the costs of the various operations that best fit the input data (In our application we also assume that optimal alignments are given for similar sequence pairs). For a more detailed treatment of inverse sequence alignment see [2] .
SVMalign is an implementation of inverse sequence alignment that uses large-margin methods for the underlying optimization, as described in [1]. It uses a simple model for sequence alignment with four distinct operations (Match, Substitution, Insertion, and Deletion) and a number of parameters associated with each (e.g. gap opening, extension, substitution matrix). SVMalign includes, as a subroutine, the Smith-Waterman algorithm for sequence alignment.
It is based on two other software packages, which are included in the distribution below:
- SVMlight – an efficient implementation of Support Vector Machines
- SVMstruct – an API for learning complex output spaces
Those familiar with SVMlight should find that the usage of SVMalign is quite similar.
Downloading and Installing the
Distribution
The distribution is available as source-code, but has successfully been compiled with gcc on Solaris, Linux, and Windows.
First, download the archive SVMalign into an empty directory. Then open up a shell prompt and type the following:
% gunzip –c svm_align.tar.gz | tar
xvf –
Compiling and linking the program should be as simple as executing the following at a shell prompt:
% make
This will create, amongst other things, two executables: svm_align_learn and svm_align_classify. To delete all intermediate files type make tidy and to delete intermediate and executable files type make clean.
This section covers the format for inputting data to SVMalign and how to use the command line interface. For a principled introduction to the methods implemented here refer to [1].
The general command line format for the learning software is:
% svm_align_learn [options] sequence_file
model_file
The options available are as follows:
General
options:
-?
-> this help
-v [0..3] ->
verbosity level (default 1)
-y [0..3] ->
verbosity level for svm_light (default 0)
Learning options:
-c float ->
C: trade-off between training error
and margin (default 0.01)
-p [1,2] ->
L-norm to use for slack variables. Use 1 for L1-norm,
use 2 for squared slacks. (default 1)
-o [1,2] ->
Slack rescaling method to use for loss.
1: slack rescaling
2: margin rescaling
(default
1)
-l [0..] ->
Loss function to use.
0: zero/one loss
(default 0)
-i string -> model
for weight initialization.
Kernel options:
-t int
-> type of kernel function:
0: linear (default)
1: polynomial (s a*b+c)^d
2: radial basis function exp(-gamma ||a-b||^2)
3: sigmoid tanh(s a*b + c)
4: user defined kernel from kernel.h
-d int
-> parameter d in polynomial kernel
-g float ->
parameter gamma in rbf kernel
-s float ->
parameter s in sigmoid/poly kernel
-r float ->
parameter c in sigmoid/poly kernel
-u string ->
parameter of user defined kernel
Optimization options (see [1]):
-q [2..] ->
maximum size of QP-subproblems (default 10)
-n [2..q] -> number
of new variables entering the working set
in
each iteration (default n = q). Set n<q to prevent
zig-zagging.
-m [5..] ->
size of cache for kernel evaluations in MB (default 40)
The larger the faster...
-e float ->
eps: Allow that error for termination criterion
(default 0.01)
-h [5..] ->
number of iterations a variable needs to be
optimal before considered for shrinking (default 100)
-k [1..] ->
number of new constraints to accumulate before
recomputing the QP solution (default 100)
Output options:
-a string -> write
all alphas to this file after learning
(in the same order as in the training set)
Structure learning
options:
--s [0,1] -> Weight of
substitution matrix (default 1)
--a int -> Manually specify the size of
the alphabet. Do this
if not all characters appear in training set. A value of 0
sets
the alphabet size to the greates value appearing in the
training set. (default 0)
--i [0-2] -> Individual
gap costs
0: Only use general, non-character-specific gap cost (default)
1: Use individual gap costs in addition to general gap cost
2: Only use individual gap costs.
--g [0,1] -> Gap
extension off/on (default 0).
--h [0,1] -> Use separate
scores for insert and delete (default 0)
Here is an explanation of the various options specific to learning alignments. For information on the other options see SVMstruct.
Substitution Matrix Weight (--s)
This essentially allows you to change the ‘importance’ of the substitution matrix. As this values approaches zero, the optimization algorithm relies less on the substitution matrix. With a value of zero the substitution matrix is not used.
Alphabet Size (--a)
By default, SVMalign sets the alphabet size to the maximum ‘character’ that appears in the sequence file. In the event that your training set does not include every character in your alphabet then it is useful to specify that value manually. However, any parameters specific to those missing characters will be 0. (A possibility for the future is to include some kind of smoothing to avoid this).
Individual Gap Costs (--i) and Gap Extension
(--g)
Here you can specify which types of gap costs to use (see Alignment Model section). The –i option is overridden if gap extension is turned on.
The general command line format for the classification software is:
svm_align_classify [options] sequence_file model_file
output_file
and the options are:
-h
-> this help
-v
[0..3] -> verbosity level
(default 2)
-f
[0,1] -> 0: output
alignments(default)
-> 1: output the margin of the alignment
The sequence_file contains the sequences to test the model contained in model_file. With the –f option at the default value, the optimal alignments for each Native-Homolog pair are printed to the output_file.
The program reports Delta Error and Tau Error, as they are described in [1].
Model File Format and Description of Alignment Model
Note: Model files
generated by previous versions of SVMalign are not compatible with
version available here.
The alignment model used by SVMalign is slightly more general than the standard models used for sequence alignment. We assume the user is already familiar with concepts such as substitution matrices and gap extension. For each operation (Match, Substitution, Insertion, Deletion) there are a few parameters that contribute to its cost. Below is the recurrence relation used to find the alignment and a brief English description.
The recurrence relation is made up of three matrices. The lefthand column and first row of each are initialized to zero and the rest of the matrices are filled in according to the relation. One then takes the maximum entry in any of the three matrices and backtraces from there to get the alignment. Conceptually you can think of the entry (I,j) in each matrix as the optimal score of aligning characters 1 through I in sequence 1 with characters 1 through j in sequence 2 where the final operation in the alignment is a match/substitution (L), insertion (F), or deletion (E).
|
|
|
|
|
|
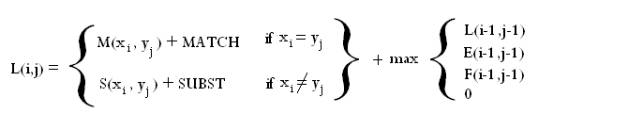
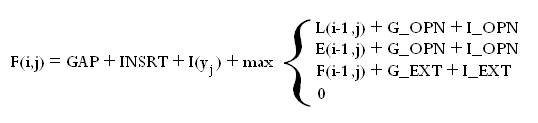
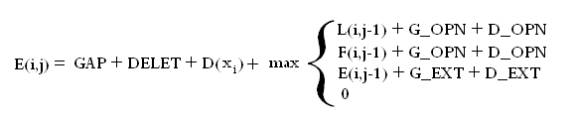
It is pretty simple to specify these parameters in the model file. Beyond the first line you are free to include any of the following, all of type double:
*Substitution Matrix Scale (SUBST SCALE): This is the weight given to the substition matrix. If no value is specified then a default value of 1 is used.
*General Match Score (MATCH): This is the amount accrued by any match occurring in the alignment.
*General Substitution Score (SUBST): This is the amount accrued by any substitution occurring in the alignment.
*General Gap Score (GAP): This is the amount accrued by any insertion or deletion occurring in the alignment:
*Gap Opening (GAP OPEN): In a series of one or more insertions or deletions this is the amount added for the first in that series. Note that a series is demarcated by any change in operation so, for instance, an insertion followed by a deletion will count as 2 Gap Openings.
*Gap Extension (GAP EXT): Counted for every insertion/deletion besides the first in a series.
*General Insert Score (INSERT): This is the amount accrued by any insertion occurring in the alignment.
*Insert Open, Insert Extension (INSERT OPEN, INSERT EXT): same as gap opening and extension except is only counted for insertions.
*General Delete Score (DELETE): This is the amount accrued by any deletion occurring in the alignment.
*Delete Open, Insert Extension (DELETE OPEN, DELETE EXT): same as gap opening and extension except is only counted for deletions.
*Individual Match Score (M(i)): This is the score for matching i.
*Individual Substitution Score (S(i, j)): This is the score for substituting i with j.
*Individual Deletion Score (D(i)): This is the score for deleting i.
*Individual Insertion
Score (I(i)): This is the
score for inserting i.
The first line of the file, however, must specify the size of your alphabet (e.g. number of words, amino acids). The line appears as:
ALPHABETSIZE: [positive integer]
As an example, let's say you want an alphabet size of 4, to scale the substitution matrix by 1/2 and all matches to cost 2.8 except for matching 1, which should cost 4.8. Then your model file would look like:
ALPHABETSIZE: 4
SUBST SCALE: .5
MATCH: 2.8
M(1): 2.0
The file sequence_file contains the training data for the program, in the form of numeric sequences. The input sequences can be thought of as grouped into ‘bundles’. Each ‘bundle’ contains the following information:
- ID
- Native sequence (s)
- Homolog sequence (+) and its alignment
- Decoy sequences (-)
A simple input file, example.dat, is included in the archive. It is easiest to look at an example to explain the formatting:
s id:21
seq:1.2.3.4.0
+ id:21
seq:1.5.3.6.0
align:1.3.4.1.2.0
- id:21
seq:9.9.9.9.9.0
- id:21
seq:8.8.8.0
The first line specifies the Native sequence for ‘bundle’ 21. The actual sequence is 1 2 3 4, but NOTE: each ‘character’ is separated by a period and is terminated by a 0. So clearly, your alphabet cannot include 0 as a character. The second line specifies a Homolog sequence for ‘bundle’ 21 in much the same way. The third line gives the sequence of operations aligning the Homolog with the Native. NOTE: The alignment line must have a tab at its beginning, not an equivalent number of spaces, and must immediately follow a line specifying a Homolog. SVMalign uses the following convention to specify various operations:
- Match
- Substitution
- Deletion
- Insertion
So in the above example the sequence of operations would be: M,D,I,M,S. The fourth and fifth lines specify decoy sequences, or sequences that should not have a high scoring alignment with the Native sequence. Every bundle can include an arbitrary number of decoys, but only one Native and Homolog. Various parts of a ‘bundle’ can appear in any order and interleaved with parts of other ‘bundles’.
[1]
T. Joachims,
Learning to Align Sequences: A Maximum-Margin Approach, Technical Report,
August, 2003.
[2]
Fangting Sun, D.
Fernandez-Baca, and Wei Yu. Inverse
parametric sequence alignment. In International Computing and Combinatorics
Conference (COCOON), 2002.
Updates
July 29, 2004 – Some bugs and memory leaks fixed. Changed model file format and alignment model. More options. New API.
Please report any questions, comments, bug reports to Philip Zigoris <paz3 (a) cs . cornell . edu>